For much of modern medicine’s history, inflammation was treated like a bit player: important in infections and injuries, but largely irrelevant to the slow, ordinary business of growing old. Aging was thought to be a matter of wear and tear, genetic fate, or accumulated bad luck. That view has collapsed. Today, chronic inflammation stands revealed not as a side effect of aging, but as one of its central engines.
This persistent, low-grade immune activation—now widely known as inflammaging—is neither dramatic nor obvious. There is no swelling, no fever, no sirens. Instead, there is a steady biochemical hiss: cytokines circulating a little too high, immune cells a little too irritable, tissues quietly stressed for decades. Over time, this background noise becomes destructive. It stiffens arteries, weakens muscles, clouds cognition, destabilizes genomes, and prepares fertile ground for cancer.
The elevation of chronic inflammation to a formal hallmark of aging in the 2023 update by López-Otín and colleagues marked a conceptual turning point. It acknowledged what decades of data had been whispering: if aging is a network of reinforcing failures, inflammation is one of the most powerful nodes in that network.
From Emergency Response to Systemic Malfunction
Inflammation evolved as a survival mechanism. When pathogens invade or tissues are damaged, inflammatory signaling rapidly mobilizes immune cells, clears debris, and initiates repair. In youth, this response is tightly regulated. It turns on when needed and shuts off when the threat is resolved.
Aging breaks this discipline.

As tissues accumulate DNA damage, mitochondrial dysfunction, senescent cells, and misfolded proteins, the immune system increasingly mistakes internal debris for external threats. Pattern-recognition pathways—such as NF-κB (a master inflammatory transcription factor), the NLRP3 inflammasome (an intracellular danger sensor), and the cGAS-STING DNA surveillance system—remain chronically engaged.
What was once a fire brigade becomes a faulty alarm system.
NF-κB exemplifies this shift. In young organisms, it is activated briefly in response to stress. With age, it becomes persistently active, driving continuous production of IL-6, IL-1β, and TNF-α. These cytokines do more than signal inflammation; they actively reshape cellular behavior. They accelerate telomere erosion, suppress autophagy, disrupt insulin signaling, and lock damaged cells into senescence. Instead of coordinating repair, the inflammatory response becomes self-reinforcing.
The NLRP3 inflammasome adds fuel to the fire. Activated by mitochondrial debris, extracellular ATP, cholesterol crystals, and misfolded proteins, it processes IL-1β and IL-18 into their active forms. Critically, NLRP3 does not require infection. It is perfectly capable of triggering sterile inflammation, where the immune system wages war against the body’s own molecular refuse.
Animal studies leave little room for ambiguity. Mice lacking NLRP3 age more slowly, maintain stronger muscles and bones, show better metabolic health, preserve cognition, and live significantly longer. This is not marginal biology. It is system-level control.
Senescence: When Retired Cells Refuse to Leave
Aging tissues accumulate senescent cells—cells that have permanently exited the cell cycle due to damage but remain metabolically active. Senescence is meant to be protective, preventing damaged cells from becoming cancerous. The problem arises when senescent cells linger.
These cells secrete the senescence-associated secretory phenotype (SASP), a toxic mixture of cytokines, chemokines, proteases, and growth factors. The SASP is inflammatory by design. It recruits immune cells, degrades tissue structure, and—most insidiously—induces senescence in neighboring cells. One senescent cell can corrupt an entire microenvironment.
NF-κB again plays a central role, orchestrating SASP expression. Over time, tissues become studded with inflammatory signal generators that never turn off. This helps explain why clearing senescent cells with senolytic drugs produces such dramatic improvements in animal models: vascular elasticity returns, physical function improves, and inflammatory markers fall.
Senescence and inflammation form a closed loop. Inflammation creates senescent cells. Senescent cells amplify inflammation. Aging accelerates.
Immunosenescence: A Paradoxical Collapse
The aging immune system fails in a peculiar way. It becomes both hyperactive and ineffective.
Innate immune cells such as macrophages and neutrophils become trigger-happy, releasing reactive oxygen species and cytokines in response to minimal stimuli. Pattern-recognition receptors grow hypersensitive. At the same time, adaptive immunity erodes. Naïve T and B cells decline, memory cells become exhausted, and immune diversity shrinks.
The result is an immune system that overreacts to debris but underperforms against real threats. This explains why older adults experience chronic inflammation alongside poor vaccine responses and higher infection mortality. It also helps explain the catastrophic inflammatory reactions seen in some elderly patients during infections like influenza or COVID-19.
Inflammaging is not immune strength. It is immune misfiring.
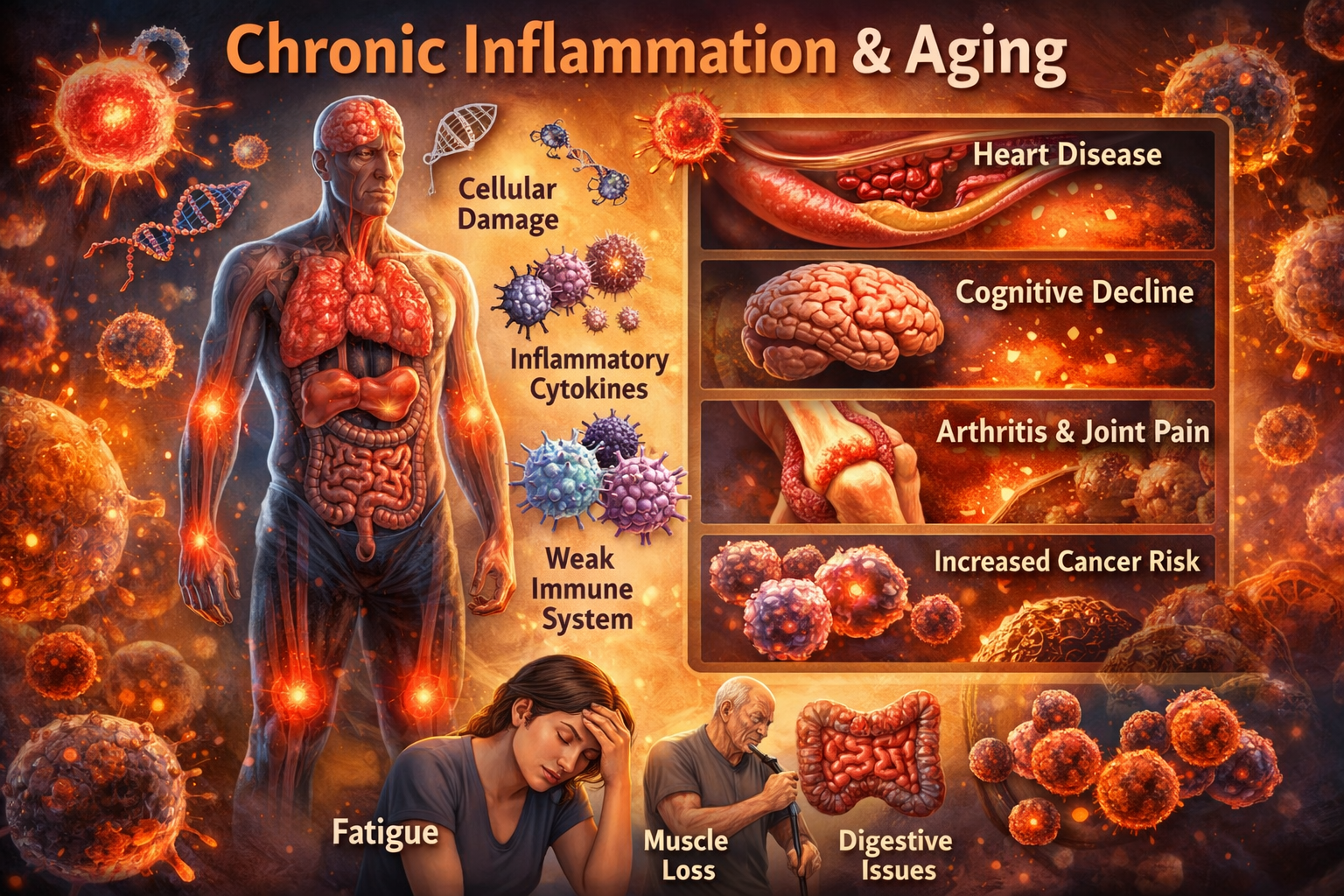
Systemic Consequences: Aging Written Across Organs

Chronic inflammation does not stay local. Cytokines circulate. Immune cells infiltrate. Endothelial barriers weaken. Over decades, nearly every organ system bears the scars.
In the cardiovascular system, inflammation drives atherosclerosis from initiation to rupture. Cytokines transform blood vessels into adhesive surfaces, recruit monocytes, promote foam cell formation, and destabilize plaques. The CANTOS trial provided definitive human evidence that suppressing inflammation—without altering cholesterol—reduces heart attacks and strokes.
In metabolic tissues, inflammation sabotages insulin signaling, accelerates muscle breakdown, and inflames visceral fat. Sarcopenia, frailty, and type 2 diabetes are not merely metabolic failures; they are inflammatory syndromes.
In the brain, peripheral inflammation primes microglia into a chronic reactive state. Blood-brain barrier integrity declines. Neuroinflammation accelerates cognitive aging and contributes to Alzheimer’s and Parkinson’s pathology. Even mood disorders in later life show inflammatory fingerprints.
In bone and joint tissues, inflammatory cytokines tilt the balance toward destruction. Osteoporosis and osteoarthritis are increasingly understood as inflammatory degenerations layered on mechanical stress.
Cancer, too, is inseparable from chronic inflammation. Inflammatory mediators damage DNA, impair repair mechanisms, promote proliferative signaling, reshape tissue architecture, suppress antitumor immunity, and create environments where malignant clones thrive. Roughly a quarter of cancers are directly linked to chronic inflammatory states, and age-related sterile inflammation likely expands that fraction.
Inflammation as a Network Hub of Aging
Perhaps the most profound insight to emerge from aging biology is that chronic inflammation does not act alone. It interacts with—and amplifies—nearly every other hallmark of aging.
It accelerates genomic instability through oxidative damage.
It hastens telomere attrition by inhibiting telomerase.
It reshapes the epigenome, locking in inflammatory gene expression.
It disrupts proteostasis and suppresses autophagy.
It worsens mitochondrial dysfunction, which feeds back into immune activation.
It exhausts stem cell pools by forcing unnecessary proliferation.
It corrupts intercellular communication across endocrine, immune, neural, and microbial systems.
Inflammation is not one strand in the aging web. It is a central tension cable.
Therapeutic Implications: From Blunt Suppression to Precision Control
The recognition of inflammaging has transformed therapeutic ambition. Instead of treating age-related diseases one by one, researchers are targeting shared inflammatory drivers.
Biologic cytokine inhibitors (against IL-1β, IL-6, TNF-α) have proven the principle but carry infection risks and high costs. NLRP3 inflammasome inhibitors offer a more upstream strategy. Senolytics aim to remove the cellular sources of inflammatory noise altogether.
Metabolic modulators such as metformin and NAD⁺ boosters dampen inflammation indirectly by restoring cellular resilience. Microbiome interventions seek to seal the leaky gut-immune axis that fuels systemic inflammation.
Equally important are lifestyle interventions, which remain the most powerful and safest anti-inflammatory tools available. Exercise lowers IL-6, CRP, and TNF-α while increasing anti-inflammatory myokines. Anti-inflammatory diets reshape immune tone through fats, fiber, and polyphenols. Caloric restriction and intermittent fasting activate ancient stress-response pathways that suppress inflammation. Sleep, stress management, and smoking cessation quietly but profoundly reduce inflammatory burden.
These interventions do not merely add benefits. They compound.
Looking Forward: Measuring and Modifying Inflammation Age
The future of anti-aging medicine will likely involve routine measurement of inflammatory burden—not just CRP, but multi-marker panels and transcriptomic signatures. Concepts like “inflammatory age” or iAge may guide early intervention, long before disease manifests.
Rather than suppressing immunity, the goal will be recalibration: restoring the ability to resolve inflammation efficiently without leaving defenses down. Specialized pro-resolving mediators, intermittent dosing strategies, and combination therapies may allow just that.
The Takeaway
Chronic inflammation is no longer an afterthought in aging biology. It is one of aging’s principal drivers and one of its most actionable vulnerabilities. By understanding inflammaging not as noise but as signal—an emergent property of damaged systems talking too loudly—we gain leverage over the aging process itself.
Aging may be inevitable. Inflammaging may not be.
The quiet fire that once seemed unavoidable is increasingly being understood, measured, and—at last—challenged.
Below is a practical, evidence-based reference guide to the main medications and supplements used to manage chronic inflammation, organized for clarity rather than hype. This is not a prescription, but a decision-support map that clinicians, researchers, and informed individuals commonly use when thinking in terms of risk–benefit–time horizon.
I’ll separate pharmaceuticals and supplements, then end with how to combine them intelligently.
————————
I. Medications for Chronic Inflammation
(Prescription or medical supervision strongly advised)
1. Low-Dose Aspirin
Mechanism: COX inhibition → ↓ prostaglandins and thromboxane
Best for: Vascular inflammation, secondary cardiovascular prevention
- Dose: 75–100 mg daily
- Benefits:
-
- Lowers CRP (C-reactive protein, a systemic inflammation marker)
- Reduces platelet activation
- Possible colorectal cancer risk reduction (long-term)
- Side effects:
-
- GI bleeding
- Hemorrhagic stroke risk
- Time frame: Weeks to months
- Notes: Not recommended for primary prevention in healthy elderly due to bleeding risk
————————
2. Colchicine (Low Dose)
Mechanism: Microtubule inhibition → ↓ NLRP3 inflammasome activation
- Dose: 0.5 mg once daily
- Benefits:
-
- Reduces IL-1β and IL-6
- Strong evidence in cardiovascular inflammation
- Useful in gout, pericarditis
- Side effects:
-
- GI upset (diarrhea)
- Rare myopathy (↑ risk with statins)
- Time frame: Days to weeks
- Evidence highlight: Reduces heart attacks independent of cholesterol
————————
3. Statins (e.g., Atorvastatin, Rosuvastatin)
Mechanism: ↓ LDL + pleiotropic anti-inflammatory effects
- Dose:
-
- Atorvastatin 10–40 mg
- Rosuvastatin 5–20 mg
- Benefits:
-
- Lowers CRP independent of cholesterol
- Stabilizes atherosclerotic plaques
- Side effects:
-
- Muscle pain
- Rare liver enzyme elevation
- Time frame: 4–12 weeks
- Note: Often underestimated as anti-inflammatory drugs
————————
4. Metformin
Mechanism: AMPK activation → ↓ NF-κB, ↓ inflammasome signaling
- Dose: 500–2000 mg/day (divided)
- Benefits:
-
- Lowers systemic inflammation
- Improves insulin sensitivity
- Associated with reduced cancer risk
- Side effects:
-
- GI upset
- Rare lactic acidosis (renal disease)
- Time frame: Weeks to months
- Special note: Being tested as an anti-aging drug (TAME trial)
————————
5. IL-1 / IL-6 / TNF-α Biologics
(e.g., Anakinra, Canakinumab, Tocilizumab, Etanercept)
- Mechanism: Cytokine neutralization
- Benefits:
-
- Powerful reduction of inflammation
- Proven cardiovascular and autoimmune benefits
- Side effects:
-
- Infection risk
- High cost
- Time frame: Days to weeks
- Use case: Severe inflammatory disease, not routine aging
————————
II. Supplements with Anti-Inflammatory Evidence
(Lower risk, slower but cumulative effects)
1. Omega-3 Fatty Acids (EPA/DHA)
Mechanism: Shift eicosanoids toward resolution (resolvins, protectins)
- Dose: 1–3 g/day combined EPA+DHA
- Benefits:
-
- Lowers CRP, IL-6
- Cardiovascular and brain protection
- Side effects:
-
- Mild GI upset
- Bleeding risk at very high doses
- Time frame: 4–12 weeks
————————
2. Curcumin (with Piperine)
Mechanism: NF-κB inhibition
- Dose: 500–1500 mg/day (bioavailable form)
- Benefits:
-
- Reduces joint and gut inflammation
- Antioxidant
- Side effects:
-
- GI discomfort at high doses
- Time frame: 2–8 weeks
- Note: Absorption is critical—standard turmeric is weak
————————
3. Magnesium (Glycinate or Threonate)
Mechanism: ↓ NF-κB activation, improves mitochondrial function
- Dose: 200–400 mg/day
- Benefits:
-
- Lowers CRP
- Improves sleep and stress (secondary inflammation reducers)
- Side effects:
-
- Diarrhea (oxide form)
- Time frame: 2–4 weeks
————————
4. Vitamin D3
Mechanism: Immune modulation, ↓ pro-inflammatory cytokines
- Dose: 1000–4000 IU/day (target 25-OH-D: 30–50 ng/mL)
- Benefits:
-
- Reduces IL-6, TNF-α
- Improves immune balance
- Side effects:
-
- Hypercalcemia at excessive doses
- Time frame: 1–3 months
————————
5. Resveratrol
Mechanism: SIRT1 activation → ↓ NF-κB
- Dose: 250–500 mg/day
- Benefits:
-
- Anti-inflammatory
- Supports mitochondrial health
- Side effects:
-
- GI upset
- Time frame: 4–8 weeks
- Evidence: Mixed but mechanistically strong
————————
6. Probiotics / Prebiotics
Mechanism: Gut barrier repair → ↓ endotoxemia
- Dose:
-
- Probiotics: ≥10⁹ CFU/day
- Fiber: 25–40 g/day
- Benefits:
-
- Lowers systemic inflammation
- Improves insulin sensitivity
- Side effects:
-
- Bloating initially
- Time frame: 4–12 weeks
————————
7. Fisetin (Senotherapeutic)
Mechanism: Senescent cell clearance → ↓ SASP
- Dose:
-
- Daily low dose: 100–200 mg
- Intermittent high dose (research): 20 mg/kg for 2 days/month
- Benefits:
-
- Reduces inflammatory burden at source
- Side effects:
-
- Limited human data
- Time frame: Months
————————
III. Practical Combination Strategy (Conceptual)
Low-risk foundation (long-term):
- Omega-3
- Vitamin D
- Magnesium
- Fiber + probiotics
- Exercise + sleep optimization
Metabolic / aging layer:
- Metformin
- Curcumin
- Resveratrol
High-risk / high-reward (medical):
- Statins
- Low-dose colchicine
- Cytokine inhibitors (select cases)
————————
IV. Expected Timeline of Improvement
- Days–2 weeks: Colchicine, NSAIDs (symptom relief)
- 4–8 weeks: Omega-3, curcumin, statins
- 2–6 months: Metformin, weight loss, exercise
- 6–12 months: Senolytics, microbiome remodeling
————————
Final Perspective
Chronic inflammation is not a single target. It is a network failure.
The most effective strategies are layered, patient-specific, and measured over time.
If you want, next we can:
- Design a personalized anti-inflammaging protocol
- Build a clinician-friendly table or printable PDF
- Map interventions to specific biomarkers (CRP, IL-6, TNF-α, iAge)
The fire can be managed—but only if you cool the system, not just the flames.
Note: This article is for educational purposes, consult your doctor or health advisor if you have any health issue.
Discover more from RETHINK! SEEK THE BRIGHT SIDE
Subscribe to get the latest posts sent to your email.